hyraxAbif: Modules for parsing, generating and manipulating AB1 files.
This is a package candidate release! Here you can preview how this package release will appear once published to the main package index (which can be accomplished via the 'maintain' link below). Please note that once a package has been published to the main package index it cannot be undone! Please consult the package uploading documentation for more information.
This library provides functionality for parsing, modifying, writing and generating ABIF files

Any AB1 file conforming to the standard at http://www6.appliedbiosystems.com/support/software_community/ABIF_File_Format.pdf should be supported.
This library also support generating a minimal ABIF file from a FASTA input sequence.
A basic terminal application that can dump and generate AB1s is included. See https://github.com/hyraxbio/hyraxAbif/blob/master/app/Main.hs
See
Hyrax.Abif.Generate for generate ABIF files from FASTA inputs
Hyrax.Abif.Read for parsing ABIF files
Hyrax.Abif.Write for creating/updating ABIF files
Hyrax.Abif for the core types
Examples for examples
[Skip to Readme]
Properties
| Versions | 0.2.3.2, 0.2.3.3, 0.2.3.4, 0.2.3.4, 0.2.3.5, 0.2.3.7, 0.2.3.8, 0.2.3.9, 0.2.3.10, 0.2.3.14, 0.2.3.15, 0.2.3.21, 0.2.3.26, 0.2.3.27, 0.2.4.2, 0.2.4.4, 0.2.4.5 |
|---|---|
| Change log | None available |
| Dependencies | base (>=4.9.1.0 && <5), binary (>=0.8.5.1 && <0.8.6.0), bytestring (>=0.10.8.2 && <0.10.9.0), directory (>=1.3.0.2 && <1.3.2.0), filepath (>=1.4.1.2 && <1.4.2.0), hscolour (>=1.24.4 && <1.25.0), hyraxAbif, pretty-show (>=1.6.16 && <1.7.0), protolude (>=0.2.2 && <0.2.3), text (>=1.2.3.0 && <1.2.4.0) [details] |
| License | BSD-3-Clause |
| Copyright | 2018 HyraxBio |
| Author | HyraxBio |
| Maintainer | andre@hyraxbio.co.za, andre@andrevdm.com |
| Category | Bioinformatics |
| Home page | https://github.com/hyraxbio/hyraxAbif/#readme |
| Source repo | head: git clone https://github.com/hyraxbio/hyraxAbif |
| Uploaded | by andrevdm at 2018-07-10T08:10:59Z |
Modules
[Index]
Downloads
- hyraxAbif-0.2.3.4.tar.gz [browse] (Cabal source package)
- Package description (as included in the package)
Maintainer's Corner
Package maintainers
For package maintainers and hackage trustees
Readme for hyraxAbif-0.2.3.4
[back to package description]HyraxBio AB1 parser, writer and generator (beta 0.2)
This project contains
- Modules for parsing, generating or manipulating AB1 files.
- Support for generating a minimal AB1 file from a FASTA input file
- A simple terminal app to perform these operations
See
- https://hackage.haskell.org/package/hyraxAbif for the hackage documentation
- http://www6.appliedbiosystems.com/support/software_community/ABIF_File_Format.pdf for a high level overview of the AB1 file format.
Building
Build with one of
stack buildor (make build)cabal new-build
Terminal app
Run with
stack exec hyraxAbif-exe -- -- dumpif you are using stackcabal new-run hyraxAbif-exe dumpif you are using cabal 2.x
Dump AB1
To dump an existing AB1 run
hyraxAbif-exe dump example.ab1
This will output the structure of the AB1 like this
Header { hName = "ABIF" , hVersion = 101 }
Directory
{ dTagName = "tdir"
, dTagNum = 1
, dElemTypeCode = 1023
, dElemTypeDesc = "root"
, dElemType = ElemRoot
, dElemSize = 28
, dElemNum = 13
, dDataSize = 364
, dDataOffset = 61980
, dData = ""
, dDataDebug = []
}
[ Directory
{ dTagName = "DATA"
, dTagNum = 9
, dElemTypeCode = 4
, dElemTypeDesc = "short"
, dElemType = ElemShort
, dElemSize = 2
, dElemNum = 7440
, dDataSize = 14880
, dDataOffset = 128
, dData = ""
, dDataDebug = []
}
.
.
.
DATA {short} tagNum=9 size=2 count=7440 offset=128 []
DATA {short} tagNum=10 size=2 count=7440 offset=15008 []
DATA {short} tagNum=11 size=2 count=7440 offset=29888 []
DATA {short} tagNum=12 size=2 count=7440 offset=44768 []
FWO_ {char} tagNum=1 size=1 count=4 offset=1195463747 ["GATC"]
LANE {short} tagNum=1 size=2 count=1 offset=65536 ["1"]
PBAS {char} tagNum=1 size=1 count=744 offset=59648 ["GGGGGCAACTAAAGGAAGCTCTATTAGATACAGGAGCAGATGATACAGTATTAGAAGAAATGAGTTTGCCAGGAAGATGGAAACCAAAAATGATAGGGGGAATTGGAGGTTTTATCAAAGTAAGACAGTATGATCAGATACTCATAGAAATCTGTGGACATAAAGCTATAGGTACAGTATTAGTAGGACCTACACCTGTCAACATAATTGGAAGAAATCTGTTGACTCAGATTGGTTGCACTTTAAATTTTCCCATTAGCCCTATTGAGACTGTACCAGTAAAATTAAAGCCAGGAATGGATGGCCCAAAAGTTAAACAATGGCCATTGACAGAAGAAAAAATAAAAGCATTAGTAGAAATTTGTACAGAGATGGAAAAGGAAGGGAAAATTTCAAAAATTGGGCCTGAAAATCCATACAATACTCCAGTATTTGCCATAAAGAAAAAAGACAGTACTAAATGGAGAAAATTAGTAGATTTCAGAGAACTTAATAAGAGAACTCAAGACTTCTGGGAAGTTCAATTAGGAATACCACATCCCGCAGGGTTAAAAAAGAAAAAATCAGTAACAGTACTGGATGTGGGTGATGCATATTTTTCAGTTCCCTTAGATGAAGACTTCAGGAAGTATACTGCATTTACCATACCTAGTATAAACAATGAGACACCAGGGATTAGATATCAGTACAATGTGCTTCCACAGGGATGGAAAGGATCACCAGCAATATTCCAAAGTAGCATGA"]
PDMF {pString} tagNum=1 size=1 count=23 offset=60392 ["KB_3500_POP7_BDTv3.mob"]
PDMF {pString} tagNum=2 size=1 count=23 offset=60415 ["KB_3500_POP7_BDTv3.mob"]
PLOC {short} tagNum=1 size=2 count=744 offset=60438 []
S/N% {short} tagNum=1 size=2 count=4 offset=61926 []
SMPL {pString} tagNum=1 size=1 count=10 offset=61934 ["S17-SeqF1"]
CMNT {pString} tagNum=1 size=1 count=1 offset=61944 ["Generated by HyraxBio AB1 generator"]
The data is output twice. The first section is the detail, the second is the summary.
Selected data types have the "debug data" element populated. e.g. the PBAS (FASTA)
Generate minimal AB1s from FASTAs
To create an AB1 run
hyraxAbif-exe gen "./pathContainingFastas" "./pathForOutputAb1s"
This will create an AB1 per input FASTA
Input FASTA format
Each input data should have the following format
> weight
read
> weight
read
-
The weight is a numeric value between 0 and 1 that specifies the weight of the current read. No other header/name is allowed
-
The read is the set of input nucleotides, IUPAC ambiguity codes are supported (MRWSYKVHDBNX). A read can be single or multi-line
Weighted reads
- The weigh of a read specifies the intensity of the peak from 0 to 1.
- Weights for each position are added to a maximum of 1 per nucleotide
- You can use
_as a "blank" nucleotide, in which only the nucleotides from other reads will be considered
For example
> 0.5
ACG
> 0.3
AAAA
> 1
__AC
Results in the following weighted nucleotide per position
- 0:
A(0.5 + 0.3) - 1:
C(0.5),A(0.3) - 2:
G(0.5),A(0.3 + 1 = 1) - 3:
A(0.3),C(1)
Note that the reads do not need to be the same length.
Example FASTA - single file
eg1.fasta
> 1
ACTG

Here there is a single FASTA with a single read with a weigh of 1 (100%). The chromatogram for this AB1 shows perfect traces for the input ACTG nucleotides
Example FASTA - two FASTA files
eg1.fasta
> 1
ACAG
eg2.fasta
> 1
ACTG
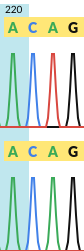
Two input FASTA files both with a weigh of 1. You can see in the second trace that the third nucleotide is a T (the trace is green). Exactly what the base-calling software (phred & recall etc) decide to call the base as depends on your settings and software choices.
Example FASTA - two FASTA files with different weights
eg1.fasta
> 1
ACAG
eg2.fasta
> 0.3
ACTG

Here the second fasta has a weight of 0.3 and you can see the traces are 30% of the height of the top ones.
Example FASTA - single FASTA with a mix
eg1.fasta
> 1
ACAG
> 0.3
ACTG

The single input FASTA has an AT mix at the third nucleotide. The first read has a weight of 1 and the second a weight of 0.3.
Notice that the maximum weight is 1, e.g. the first A has the same intensity as the second even though the first one has the reads weighted both 1 and 0.3
Example FASTA - Multiple mixes
eg1.fasta
> 1
ACAG
> 0.3
_GT
> 0.2
_G

Using the modules
- Hyrax.Abif: The core AB1 types
- Hyrax.Abif.Fasta: A simple FASTA parser used when generating AB1s
- Hyrax.Abif.Read: Module for parsing an existing AB1
- Hyrax.Abif.Write: Module for writing a new AB1 file
- Hyrax.Abif.Generate: Module for generating a minimal AB1 from a given FASTA input
For a detailed overview of the code see TODO and the haddock documentation TODO
For now the terminal app (Main.hs) serves as an example and the best starting point to understand the code
E.g. Add a comment to an existing AB1 file
import qualified Hyrax.Abif as H
import qualified Hyrax.Abif.Read as H
import qualified Hyrax.Abif.Write as H
addComment :: IO ()
addComment = do
abif' <- H.readAbif "example.ab1"
case abif' of
Left e -> putText $ "error reading ABIF: " <> e
Right abif -> do
let modified = H.addDirectory abif $ H.mkComment "new comment"
H.writeAbif "example.modified.ab1" modified
For additional examples see the Examples directory